克隆与转化
克隆概述
传统的克隆实验包括以下几种操作:将DNA 片段插入到质粒中,以修饰或引入抗性基因、启动子和信号序列或开放阅读框等,用于基因表达或下游蛋白表达研究。在开始克隆实验之前定出克隆策略是很重要的。需要考虑的问题有:酶切后是否有与目标载体匹配的末端,插入序列在载体上可能的方向,以及插入序列是否在读码框内,是否会影响翻译。以下指南有助于克隆的设计和相关问题的解决。
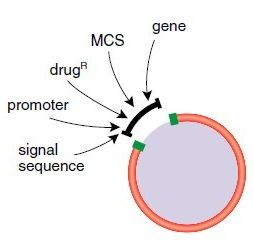
准备插入片段和质粒
来源于质粒的插入片段
• 选择适当的限制性内切酶切割质粒,获得能直接克隆于载体的 DNA 片段。选择产生不同末端的两种限制性内切酶来进行单向克隆。关于优化限制性内切酶反应的指导和问题解决建议,见 2013﹒2014 商品目录
275-277 页或 NEB网站的技术参考部分。
来源于
PCR 的插入片段
• 引物设计时引入能单向克隆到载体的限制位点
• 大多数限制性内切酶的识别位点上游需要 6 个保护碱基
• 如需高保真扩增可以选择具有校读功能的聚合酶,如:Q5® 超保真DNA 聚合酶(NEB
#M0491)
• PCR 优化指南和问题解决见 2013﹒2014 商品目录 325-326
页或 NEB 网站上技术参考部分
• PCR 产物可通过凝胶电泳回收或离心柱纯化
• 选择用适当的限制性内切酶消化
来源于退火寡核苷酸的插入片段
• 退火的寡核苷酸用来引入 DNA 片段(如启动子和聚合接头等)
• 两条带有互补的 5´ 或 3´ 突出末端寡核苷酸退火后能被连接到用适合的限制性内切酶切割的载体中
• 未磷酸化的寡核苷酸用 T4 PNK (NEB #M0201)
常规退火反应
寡核苷酸
5 µl (10 µM 储液)
10X 连接缓冲液
5 µl
总体积
至 50 µl
温度 85℃
10 分钟,缓慢降温(30-60 分钟)
载体
• 选择适当的限制性内切酶消化载体,最好使用能产生不匹配末端的内切酶,以防载体自连。
去磷酸化
• 去磷酸化反应是用来防止自身连接。NEB 提供两种 DNA 去磷酸化的产品。
– 小牛肠碱性磷酸酶(CIP)(NEB #M0290)是一种活力较强的去磷酸化酶,在许多不同条件下及各种NEBuffer 中均有活性。然而,CIP 不能被热失活,因此需要在连接前纯化
DNA。建议 CIP 的使用量不要超过推荐用量,以达到更好的纯化效果。
– 热敏磷酸酶(AnP)(NEB #M0289)不仅具备
CIP 的所有功能而且还能被热失活。AnP 需要锌离子发挥活性
AnP 去磷酸化
AnP
1 μl(5 单位)
DNA
1-5 μg
10X 缓冲液
2 μl(终浓度 1 mM)
总体积
至 20 μl
温育
37℃ 15 分钟(5´ 突出末端/平末端)或 60 分钟(3´ 突出末端)
热失活 65℃
5 分钟
平齐化/末端修复
• 有时插入的片段和载体需要进行末端平齐化/末端修复
• 用具有校读活性的聚合酶进行 PCR,其产物的绝大部分是平末端
• T4 DNA 聚合酶(NEB #M0203)或 Klenow(NEB
#M0210)能补平 5´ 突出末端(如 EcoRI-HF),切割 3´
突出末端(如 PstI-HF)
• 快速末端平齐试剂盒(NEB #E1201)能在 30 分钟内完成
DNA 末端的平齐化和磷酸化
磷酸化
• 连接两个
DNA 片段时,至少其中一个片段的末端应含有 5´ 磷酸(插入片段或载体)
• 引物通常以非磷酸化形式提供,因此 PCR 产物的 5´ 末端为非磷酸化,限制性内切酶消化的 DNA 其 5´ 末端都有磷酸化
• DNA 片段可以通过与 T4 PNK NEB #M0201)温育进行磷酸化
T4 PNK 磷酸化
T4
PNK 1
μl(10 单位)
DNA
1–2 µg
10X T4 PNK 缓冲液
5 μl
10 mM
ATP
5 μl(终浓度 1 mM)
无核酸酶污染的水
至 50 μl
温育
37℃ 30 分钟
纯化载体和插入片段
• 连接反应前,用凝胶电泳切胶回收或离心柱回收方法纯化载体或插入片段
• β-琼脂糖酶 I(NEB #M0392)可以用于纯化低熔点琼脂糖中的 DNA,DNA 的纯化还可用离心柱及树脂法
• 用长波 UV(360 nm)观察琼脂糖凝胶电泳结果,以减少
UV 暴露造成的 DNA 损伤
连接载体和插入片段
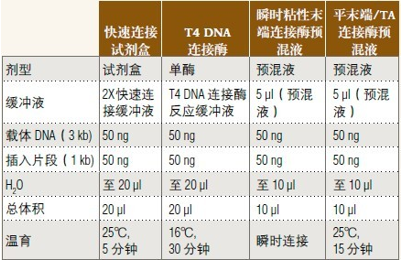
• NEB 目前提供预混液形式的连接酶,请在
2013﹒2014 商品目录第 100 页的连接酶选择表格中选择
• 载体和插入片段的摩尔比为 1:3
• 室温解冻并混匀连接缓冲液
• 平末端连接需要延长连接时间或使用高浓度的连接酶/连接酶预混液
• 连接后置于冰上并进行转化反应
• 用快速连接缓冲液或连接酶预混液时勿热失活,以免抑制转化反应
转化
• 关于感受态细胞的信息和属性,请参考2013﹒2014 商品目录第 202-213 页
• 提高感受态细胞效率的方法,请参考 2013﹒2014 商品目录第
323 页
• 通过电击的方式转化,建议您选择电转连接酶TM
用 NEB 连接酶连接
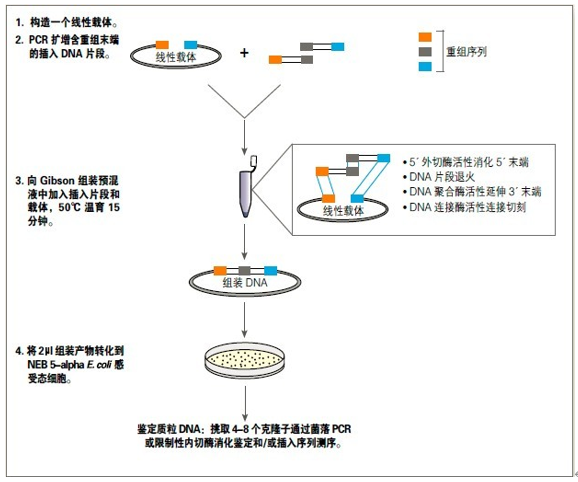
使用 Gibson 组装试剂盒克隆
Gibson 组装试剂盒中,包含 NEB
5-alpha E. coli 感受态细胞,经过优化,该试剂盒在一个试管中经等温反应,可以高效的将 1 或 2 个组装片段插入到任何载体,完成组装和克隆。更多详细信息请参考 2013﹒2014
商品目录第 127 页或登陆 NEB 网站查询。
设计引物
• 为了高效组装 PCR 片段到载体中,建议使用 15-25 个核苷酸重叠序列的引物,且其 Tm 值等于或高于
48℃
• 为了更有效的设计 Gibson 组装引物,建议您登陆
NEBGibson.com 使用在线引物设计软件 NEBuilder 设计
引物,也可以参考 Gibson 组装引物设计说明 PCR 扩增片段
• 建议使用 Q5 超保真 DNA 聚合酶(NEB #M0491)或相关产品(NEB
#M0493, NEB #M0492)扩增组装片段
• 以质粒为模板进行扩增时,建议每50μl PCR 反应体系中使用微量的 DNA(0.1–0.5
ng)进行扩增
• 通过凝胶电泳验证 PCR 产物的纯度和产量
• 如果产物纯度 > 90%,就无需对 PCR 产物进行纯化
• 在 Gibson 组装反应体系中,未纯化的 PCR 产物可以占到 20%
• 如果必须使用大量的质粒作为模板,扩增后用 DpnI(#R0176)消化
PCR 产物以降低背景
载体线性化
• 使用限制性内切酶线性化质粒时,NEB 建议使用两种限制性内切酶进行双酶切以降低未切割载体的背景
• NEB 推荐您使用 Q5 超保真 DNA 聚合酶(NEB
#M0491)或者相关产品(NEB #M0493,NEB #M0494)扩增载体
建议反应体系
& 条件
• 纯化用于组装的 DNA,可以溶于 dH2O(推荐使用 Milli-Q 水或同等纯度的水)、TE
或者其它稀释缓冲液
• 当组装 1-2 个片段到一个载体中时,NEB 推荐您使用总量为 0.02-0.5
pmols 的 DNA 片段
• 为了优化组装,在加入 Gibson 组装预混液之前调整 DNA 的体积到
10 μl
• 若 DNA 的体积大于 10 μl,相应的调整 Gibson 组装预混液的体积
• Gibson 组装试剂盒经优化可在 50℃ 15 分钟完成组装。反应时间无需超过 15
分钟,某些情况下延长时间(60 分钟)可提高效率
• 不要过夜反应
Gibson 组装克隆方法流程
转化到
NEB 5-alpha 高效 E. coli 感受态细胞
• NEB 5-alpha 高效 E. coli 感受态细胞(包含在试剂盒中)建议用于组装<
20 kb 的片段
• 大肠杆菌对有些 DNA 结构有选择拮抗,如倒置和串联重复序列,可能会导致转化效率极低或者生长极差的小克隆
• 电转化可以把转化效率提高几个数量级,因此 Gibson 组装预混液用于电击转化时,必须把反应体系稀释
3 倍,再取 1 μl 用于转化。
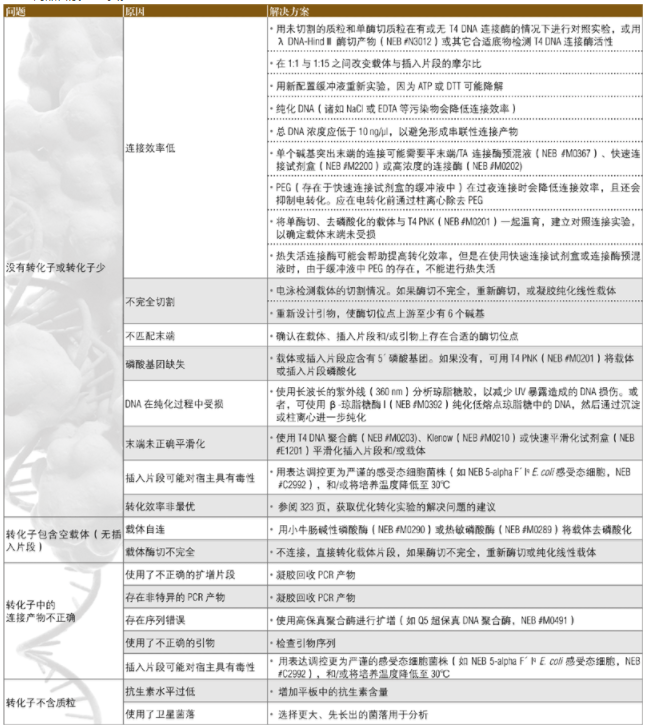
克隆问题解决指南
下面的指南可用于解决分子克隆实验中所出现的问题。关于优化转化实验的其它建议参阅 2013﹒2014 商品目录 323 页。
遗传标记
基因型反映了机体
DNA 的遗传状态,是一个理论概念。基因型决定了株系的外在特性(即表型,见下述)。在E. coli 中,一般只列出缺失基因(1),没有列出的基因,即被认为没有突变*+。原 K-12 菌株中的前噬菌体和质粒(F、λ、e14、rac)一般不列出,除非其有缺失。但是,λ存在于菌株时则被列入基因型中,所有情况下 F 因子及其变体也都列出。前噬菌体和质粒用圆括弧或方括弧标示出。基因用三个斜体的小写字母示(如dam),便于记忆且能描述该基因的功能(dam 即表示 DNA adenine
methylase,DNA 腺嘌呤甲基化酶)。
如果同一功能受几个基因调控,就用大写斜体字母来区别这几个基因(如:recA、recB、recC 和 recD 均能调控重组功能)。基因型正规的表达法应省略上标 +/-,但有时为了更清楚地反映遗传信息而并未将其省掉,比如F´ lac-proA+B+。缺失突变用 Δ 表示,并在其后用圆括弧标出缺失基因的名称
[如 Δ (lacpro ) ],标出的两基因之间的基因并未列出,但也属缺失基因。特定的突变标出其等位基因号,通常用斜体阿拉伯数字表示(如 hsdR17),而且也可用一些特殊标记来说明某突变,如 am = amber(UAG) 突变或 ts
= 高温失活。有些常见的等位基因也不一定都遵循这些原则(如:Δ (lac-pro ) X111)。如果在两种菌株的基因型中列出了带有相同等位基因号的基因,那么它们应含有完全相同的突变。
菌株的表型即为该菌株所表现出来的形态行为特性,如:Lac- 表示不能以半乳糖作为唯一碳源生长。表型用大写罗马字母表示,并总标有上标 +/- [或者 r(resistant) /s (sensitive) ]。严格地说,表型并不属于基因型范畴,但当基因型的描述并非十分具体明了时,也常将表型列于基因型之后 [如:rpsL104 (Strr)- 基因名 rps 源自 ribosomal protein, small subunit, S12,具有链霉素抗性]。
下面和下一页描述了一些常见的基因。参考文献2 收集了所有遗传定义的基因,也可登陆耶鲁大学的 E.coli Genetic Stock Center(CGSC)网站http://cgsc.biology.yale.edu/ 查看。CGSC
上的其它信息可通过发送邮件(cgsc@yale.edu)从馆长
Mary Berlyn 处获得。
*经过四十多年的研究,大多数 E. coli 实验室菌株已发生了严重的突变,且不同的株系可能存在不同的、至今尚未发现的突变,这些突变可能会也可能不会对科研实验产生影响。基于这个原因,在实验过程中,最好尝试一种以上或选用不同来源背景的菌株。
†E. coli B 及其衍生型一般是
Lon- 和 Dcm- 型,即使它们是野生型时,我们也将 Lon- 和 Dcm- 列于方括号中。
参考文献
(1) Demerec et al.
(1966) Genetics, 54, 61–76.
(2) Berlyn, M.K.B. (1996). In F. C. Niedhardt et al. (Ed.), Escherichia
coli and Salmonella:cellular and
molecular biology, (2nd ed.), Vol. 2, (pp.
1715–1902). ASM Press.
(3) Raleigh, E.A. et al. (1991) J. Bacteriol., 173, 2707–2709.
dam 缺失内源 GATC 腺嘌呤甲基化功能。Dam菌株有较高的重组频率,不断地发挥 DNA 修复功能难以转入 Dam 修饰的质粒。用于制备对某些限制性内切酶(如BclI)切割敏感的 DNA。
dcm 缺失内源 CCWGG 胞嘧啶甲基化功能。用于制备对某些限制性内切酶(如 AvaII)切割敏感的 DNA
dnaJ 一种伴侣蛋白被失活。这种缺失可以稳定 E. coli 菌株中表达的某些突变蛋白。
dut 无 dUTPase 活性。同 ung 一起可催化尿嘧啶掺入到 DNA 上。适用于某些寡核苷酸突变实验。
endA 非特异性核酸内切酶 I 活性缺失。应用 endA 菌株可制备出高质量的 DNA。
e14 一种可切割的类前噬菌体元件,存在于K-12 中,但在很多衍生菌株中丢失。e14 带有 mcrA 基因,因此e14- 菌株也属于 McrA- 型。
F 低拷贝数的自传递型质粒。F´ 因子带有部分 E. coli 染色体 DNA,特别是如 F´ lacproA+B+ 上的 lac 操纵子和 proAB。
fhuA 铁离子转运受体发生了突变,并由此获得对 T1 噬菌体的抗性(氧肟酸铁盐转运,ferric hydroxamate uptake)。以前命名为 tonA。
gyrA 在 gyrase 亚基 A DNA 上有点突变,因此,具有新霉素抗性。
hflA 增加了 λ噬菌体感染时的溶源化几率。
hsdR, 不含有特定序列甲基化的 DNA 可被
hsdS EcoKI 或 EcoBI 视为异物并被消化降解。这些酶识别不同的序列,由 hsdRMS 的不同等位基因编码。突变体去除了消化限制功能但保留了保护性甲基化功能(r-m+),而 hsdS 突变体则去除了这两种功能(r-m-)。后者制备的DNA 转入野生型菌株时将被消化掉。
提高转化效率
转化效率定义为转化
1 μg 质粒至给定体积的感受态细胞中所能产生的菌落形成单元(cfu)的数量。但是,转
化 1 μg 质粒是不常见的。实际上,转化效率常以在最适条件下转化 100 pg-1 ng 高纯度超螺旋质粒来计算。计
算转化效率(TE)的公式为:TE = 菌落数/μg/稀 得最佳实验结果。
推荐方案
高效转化方法
1. 冰上解冻细胞
10 分钟
2. 细胞中加入 1 pg-100 ng 的质粒 DNA(1-5
μl),混匀,切忌蜗旋
3. 置于冰上 30 分钟
4. 42℃ 热激 10-30 秒或根据推荐条件进行热激
5. 置于冰上 5 分钟
6. 加入 950 μl 室温的 SOC
7. 37℃ 振荡(250 rpm)培养 60 分钟
8. 混匀细胞,切忌蜗旋,并用 SOC 做系列 10 倍稀释
9. 每个稀释比例取 50-100 μl 稀释液涂布于预热的选择平板上,37℃(SHufflee® 菌株 30℃)过夜培养或按推荐条件培养
5 分钟转化方法
( 与上述方法相比只有
10% 的转化效率 )
1. 手握解冻细胞
2. 细胞中加入 1 pg-100 ng 的质粒 DNA (1-5 μl),混匀,切忌蜗旋
3. 置于冰上 2 分钟
4. 42℃ 热激 30 秒或按推荐条件进行热激
5. 置于冰上 2 分钟
6. 加入 950 μl 室温的 SOC。快速取 50-100 μl涂布于选择平板上,37℃
过夜培养(Shuffle 菌株30℃)。
注意:使用非氨苄青霉素时,可能需要在涂板前生长一定时间
转化技巧
解冻
• 细胞最好在冰上解冻
• 管中的最后一点冰融化后,立即加入 DNA
• 可以手握解冻细胞,但是一旦温度高于0℃ 时,转化效率就会下降
细胞与
DNA 冰育
• 冰育 30 分钟。预计每缩短
10 分钟会降低转化效率 2 倍
热激
• 温度和时间取决于转化体积和使用的容器,一般为 42℃ 孵育 30 秒
生长
• 37℃ 生长 1 小时对于细胞恢复和抗生素抗药性表达具有最佳效果。预计每缩短 15 分钟会有 2 倍的转化效率损失
• SOC 比 LB 培养基的转化效率高 2 倍
• 振荡或旋转晃动试管可使转化效率提高 2 倍
平板
• 平板可冷可温,可干可湿,对转化效率无明显影响
• 干温的平板更易于涂布,并实现快速菌落形成
DNA
• 用于转化的
DNA 应纯化,并重悬于水中或TE 缓冲液中
• 直接从连接混合物中取 10 μl 的 DNA 用于转化,仅有
2 倍效率损失
• 为实现最佳转化,可通过柱离心或酚/氯仿抽提和乙醇沉淀的方法纯化 DNA
• 最适 DNA 用量比通常认为的要低,纯化超螺旋 pUC19 的用量在 100 pg-1 ng 之间时转化效率最高。但是,随着 DNA 用量增加至100 ng,单次转化反应中得到的总克隆数也会增多
避免 DNA 污染

用 T7 表达菌株进行蛋白表达
T7 蛋白表达
1. 转化表达质粒至
T7 表达菌株,涂布于抗生素选择平板,37℃ 培养过夜(SHuffle® 菌株30℃ 生长 24 小时)。
2. 挑一个独立菌落至 10 ml 含抗生素的液体培养基中。
3. 37℃ 培养直至 OD600 达到
0.4-0.6。
4. 加 40 μl 100 mM IPTG 贮存液(终浓度 = 0 .4 mM),37℃ 诱导 2 小时(SHuffle
菌株 30℃ 生长 4 小时或16℃ 生长过夜)。
5. 通过考马斯亮蓝染色蛋白胶、Western Blot 或活性检测来观察表达情况。检查总细胞提取物(可溶+ 不溶的)和可溶性部分的表达情况。
6. 大规模表达时,应取新生菌落或新鲜 10 ml 培养液接种到 1 升液体培养基(含抗生素)中,37℃ 培养直至 OD600 达到
0.4-0.6。加IPTG 至终浓度为 0.4 mM,37℃ 诱导 2 小时或
15℃ 过夜(SHuffle 菌株 30℃ 生长 4 小时或
16℃ 生长过夜)。
转化反应问题解
无菌落或液体培养基中无细菌生长
• 即使 T7 表达为严格调控型,但是 T7 表达宿主菌中也可能存在低水平的基础表达。如果表达的蛋白可能有毒性,表达质粒的转化应使用下列系统:
• Iq 菌株中过量表达的 LacIq 抑制物能够降低 T7 RNA 聚合酶的基础表达。
• 在 lysY 菌株中,突变的 T7 溶解酶与
T7RNA 聚合酶相结合,从而减少目的蛋白的基础表达。诱导后,新合 成的 T7 RNA 聚合酶抵消了溶解酶作用,目的蛋白得到表达。
• 30℃ 或室温培养也可减轻毒性问题。
• 检查抗生素浓度(用对照质粒检测)。、
电泳无蛋白或无蛋白活性
• 检查毒性—细胞可能删除或缺失了表达质粒的元件。如果有毒性表达问题,试用Iq和/或 lysY 宿主以减少基础表达。
• 培养用于诱导蛋白表达的细胞。诱导前,将样品平行涂布于有抗生素和无抗生素选择标记的平板。如果有毒性产生,则两个平板菌落数会有显著的不同。在含抗生素的平板上,菌落生长数量极少(表明质粒丢失了)。
诱导蛋白不可溶
T7 表达蛋白经常产量非常高,导致目的蛋白不可溶,解决方案如下:
• 低温诱导(低温 12-15℃ 过夜)
• 降低 IPTG 浓度至 0.01 mM-0.1 mM
• 缩短诱导时间(可以缩短至 15 分钟)
• 细胞生长早期开始诱导(OD600 = 0.3 或 0.4)
转化反应问题解决指南
下面的指南可用于解决转化过程中所出现的问题。关于设计与优化克隆实验的其它建议见 2013﹒2014
商品目录 318-319 页。
